
Действие организма на лекарства. Пути и способы введения препаратов. Современные лекарственные формы и упаковки. Как отличить подделку. Лекарства-дженерики. Фармакокинетика: всасывание, распределение, превращение и выведение лекарств.
Обычно, принимая лекарство, мы не слишком задумываемся о его дальнейшей судьбе в организме. Это понятно. Нам важен результат, а не понимание того, что происходит внутри нас, когда лекарство попадает туда. А ведь, прежде чем принести облегчение, лекарство должно совершить настоящее путешествие, чтобы оказаться в нужном месте, в нужное время, да еще не растерять своего оружия. Этот путь может быть длинным или коротким, но он всегда сложен, и на каждом шагу «маленького доктора» поджидают заранее расставленные ловушки, барьеры и водовороты биохимических превращений. Давайте же мысленно попробуем проследить за каждым шагом нашего «отважного путешественника».
Наука, которая изучает взаимодействие лекарств и живых организмов, называется фармакологией, и она является частью обширного комплекса медицинских наук. Происхождение слова «фармакология» греческое: от «фармакон» — лекарство и «логос» — наука. Но еще в словаре древних египтян можно найти определение «фармаки», что в переводе звучит как «дарующие исцеление».
Лекарство — это то, что лечит, приносит облегчение при болезни или же способствует выздоровлению. Согласно этому определению, лекарством может стать и хорошая беседа, и внимание со стороны близких или незнакомых нам людей. Но для фармакологии лекарством является вещество, которое, попадая в живой организм, вызывает изменение биологических функций за счет химического или физико-химического взаимодействия. Лекарство может быть твердым, жидким или газообразным, иметь маленький или большой размер молекул, а также обладать целым рядом других физических, физико-химических и химических свойств, каждое из которых отражается на его биологическом действии. Лекарство может быть аналогом природных веществ или синтезируемых в нашем организме (например, алкалоид или гормон) или быть веществом, которое не имеет таких аналогов. Яды часто также являются лекарствами (вспомните «пчелиный яд» или «змеиный яд»), в то же время любое безопасное лекарство может стать ядом — здесь все зависит от дозы.
Модное нынче лечение травами, или фитотерапия, отнюдь не так безвредно, как это декларируют его приверженцы, призывающие отказаться от «химических лекарств» в пользу «натуральных». Самолечение в любом случае вредно, но при «самодеятельном» употреблении лекарственных трав следует учесть еще и то, что ни грамотность приготовления лекарственного средства, ни точность его дозировки (все то, что нам, кстати, гарантировано при приеме «классических» форм лекарств — таблеток, капсул и прочих) зачастую бывают просто недостижимы, и это приводит к тяжелым последствиям. Вспомним, что Джульетта впала в летаргический сон, выпив всего-навсего настойку белладонны (правда, заведомо большую дозу), применяемую обычно для снятия боли при язвенной болезни желудка и двенадцатиперстной кишки. А вот неправильно приготовленный отвар из травы сенны может привести к резким болям и спазмам в животе — не так трагично, как в предыдущем примере, но, согласитесь, тоже неприятно (особенно, если вспомнить, что готовят его для страдающих запором).
Чтобы лекарство было легче принимать и оно подействовало нужным образом, ему придают определенный вид. При этом применяют различные добавки, позволяющие получить и сохранить форму, изменить неприятный вкус, удлинить (пролонгировать) действие препарата и так далее. Созданные таким образом таблетки, капсулы, растворы, свечи, мази, пластыри называют лекарственной формой. Лекарственных форм великое множество. Условно их разделяют на четыре группы: твердые, жидкие, мягкие и газообразные. К твердым лекарственным формам относят таблетки, капсулы, порошки, гранулы, драже, брикеты и тому подобные. В эту же группу входят и всевозможные сборы, составленные из нескольких видов лекарственного растительного сырья. Жидкие формы — различные растворы, суспензии, сиропы, капли, эмульсии, настойки, экстракты. Мягкие — мази, кремы, гели, линименты, пасты, свечи, пластыри; газообразные — средства для ингаляционного наркоза, аэрозоли и так далее. Для справки в приложении 4 перечислены все применяемые в настоящее время лекарственные формы.
За последние 10-20 лет наука о лекарствах и их производство шагнули далеко вперед. Созданы новые эффективные лекарственные формы, позволяющие сократить частоту приемов, обеспечить равномерное и длительное высвобождение действующих веществ, уменьшить вероятность побочных эффектов. Применение таких форм облегчает пользование лекарствами и дает более ощутимый результат в лечении. Проконсультируйтесь с врачом или работником аптеки: они обязательно посоветуют вам лучшее из имеющихся лекарств. Даже если стоимость его окажется несколько выше, затраты несомненно окупятся удобством употребления и эффективностью воздействия.
При покупке лекарственного препарата обязательно обратите внимание на его упаковку. В последнее время участились случаи выявления подделок среди наиболее популярных лекарственных препаратов (причем, в ряде случаев отличить подделку от оригинала довольно трудно). Фармацевтические фирмы — производители лекарств, препараты которых особенно часто подделывают, принимают меры для предотвращения фальсификации. Они обращаются в информационные издания, как специализированные, так и популярные, с предупреждающими публикациями. Представители этих фирм посещают врачей и аптечных работников, информируя их о возможных подделках, объясняют, как отличить подлинные препараты от фальсифицированных. Производители постоянно усовершенствуют упаковку, вводя дополнительные степени защиты: голограммы, объемную печать, специфические шрифты и так далее. Каждая партия препарата имеет «Сертификат соответствия», который по вашей просьбе должен быть предоставлен аптечным работником.
Упаковка лекарственного препарата может быть 2-х видов: внутренняя (первичная) и внешняя (вторичная). Лекарство может иметь как оба вида упаковки, так и один. Первичная упаковка находится в непосредственном контакте с лекарственным средством. Например, таблетки могут быть упакованы в блистеры или баночки, капли или растворы — в ампулы или флаконы, мази и кремы — в баночки или тюбики (тубы) и так далее. Для предотвращения повреждений или по другой причине первичная упаковка тоже может быть упакована, например, в коробку. Это будет вторичная упаковка.
На территории Российской Федерации установлены единые требования к оформлению и маркировке лекарственных средств (Федеральный Закон РФ № 86-ФЗ от 22 июня 1999 года «О лекарственных средствах», статья 16 «Маркировка и оформление лекарственных средств»).
В качестве примера правильного оформления мы привели упаковку препарата «Куриозин» (смотри рисунок 1.2.1).
1. На первичной и вторичной упаковке хорошо читаемым шрифтом на русском языке должны быть указаны:
— название лекарственного средства и название действующего вещества (если препарат содержит 1 компонент);
— название фирмы — производителя;
— номер серии и дата изготовления;
— способ применения лекарственного средства;
— дозировка и количество доз в упаковке;
— срок годности;
— условия хранения препарата;
— условия отпуска в аптечных учреждениях (препарат выдается по рецепту врача или без);
— меры предосторожности, которые следует соблюдать при применении данного лекарства.
2. Лекарственные препараты должны поступать в продажу только с инструкцией по применению, содержащей следующие данные на русском языке:
— название и юридический адрес фирмы-производителя;
— название лекарственного средства, название действующего вещества (если препарат содержит 1 компонент);
— сведения о компонентах, входящих в состав лекарства, их дозировках, упаковке;
— сведения по фармакологическому действию активного вещества;
— показания к применению, а также противопоказания;
— возможные побочные эффекты препарата;
— возможные взаимодействия с другими лекарственными средствами;
— способ применения лекарства;
— срок годности и условия хранения;
— указание о том, что лекарственное средство следует хранить в местах, не доступных для детей;
— условия отпуска (препарат выдается по рецепту врача или без него).
3. Дополнительно на упаковке могут быть помещены следующие данные:
— логотип фирмы-производителя;
— страна-производитель;
— название препарата и действующего вещества на английском (или латинском) языке; рядом с названием может размещаться знак оригинальности, указывающий на то, что оно является торговой маркой данного производителя и не может быть использовано другим производителем;
— штрих-код.
Следует знать, что одно и то же действующее вещество (или несколько компонентов) может содержаться в одинаковых дозах в препаратах, имеющих разные торговые названия (препараты-синонимы). Поэтому, если врач или аптечный работник предложит вам заменить один препарат другим, не стоит сразу отказываться. Это может быть то же самое лекарство, но названное по-другому. Тщательно прочтите инструкцию к препарату и убедитесь в том, что препарат рекомендован вам для замены правильно.
Когда срок патентной защиты истекает, препарат переходит из разряда оригинальных в разряд дженериков (от англ. generic — родовое). Их могут производить разные фирмы, оставляя прежнее название препарата или называя по-своему (обычно это связано с разделением затрат на рекламу). Препараты-дженерики дешевле оригинальных, так как затраты на их разработку не включены в цену. Многие дженерики являются высокоэффективными препаратами и обеспечивают требуемый терапевтический эффект.
Поскольку влияние лекарства на организм не бывает односторонним, и организм тоже воздействует на лекарство, мы употребляем слово «взаимодействие». В фармакологии воздействие организма на лекарство обозначают термином фармакокинетика, а лекарства на организм — фармакодинамика.
Фармакокинетика описывает процессы, от которых зависит концентрация лекарства в организме: всасывание, распределение, биотрансформация (превращение) и выведение.
Давайте же немного приоткроем дверь в мир фармакологии, предмет которой — лекарства — интересен каждому из нас, ведь всем нам так или иначе приходится с ними соприкасаться в течение всей жизни.
Представим себе, что у нас есть препарат, который поможет избавиться от боли. Нам нужно только доставить его в кровоток. Ведь для того, чтобы лекарство дало лечебный эффект, оно должно сначала попасть в кровь. Только после этого, преодолев ряд внутренних барьеров, оно сможет дойти до цели, связаться с клетками-мишенями, вызвать нужные изменения в функционировании тканей, органов и систем (что и является проявлением его биологического действия) и, наконец, подвергнувшись превращениям (биотрансформации), или в неизмененном виде покинуть организм.
Какими путями лекарство может попасть в кровоток? Принято различать два принципиально различных способа: через желудочно-кишечный тракт (энтерально) и минуя желудочно-кишечный тракт (парентерально). Энтеральные пути введения: через рот (такой путь называют пероральным), под язык (сублингвально) и через прямую кишку (ректально). Парентеральные — на кожу и слизистые оболочки (например, вагинально, то есть на слизистую оболочку влагалища), инъекции, ингаляции. Выбор способа введения зависит от многих причин и в каждом случае определяется врачом.
Наиболее удобный и естественный для пациента путь введения — через рот — является одновременно и самым сложным для лекарства, так как оно должно преодолеть два наиболее активных внутренних барьера — кишечник и печень, где большинство веществ подвергается превращениям.
С помощью иглы лекарство можно доставить в любую точку тела, при этом обеспечены и точность дозирования, и быстрота наступления эффекта. Но это более трудоемкий способ, требующий соблюдения стерильности и присутствия медицинского персонала. Да и сам укол не настолько удобен и безболезнен для пациента, как проглатывание пилюли.
Ректальный путь введения используют, например, при заболеваниях желудочно-кишечного тракта или когда больной находится в бессознательном состоянии. Преимущество этого способа в том, что около трети лекарства поступает в общий кровоток, минуя печень.
Ингаляции применяют для прямого воздействия на бронхи или получения быстрого и сильного эффекта, так как всасывание лекарств в легких происходит очень интенсивно.
Часто, чтобы получить местный эффект, лекарство применяют наружно в виде капель в нос, глаза и уши, примочек и тому подобное.
Как видите, существуют различные пути введения препаратов: через рот, в виде инъекций, ректально, наружно; и часто один препарат имеет разные лекарственные формы. Такое многообразие — не прихоть разработчиков лекарств, а необходимость. Как правило, лекарства являются чужеродными для организма веществами, и он всяческими способами пытается нейтрализовать их и вывести наружу. На каждом шагу лекарства подвергаются воздействиям, которые могут сделать их бесполезными и даже вредными. Ведь не часто удается доставить препарат непосредственно к очагу поражения, как, например, мы это делаем, нанося мазь на воспаленный участок кожи, или закапывая раствор в больной глаз. Обычно путь лекарства в организме не прост и изобилует барьерами и препятствиями. Рассмотрим подробнее все, что происходит с лекарством на этом пути.
Введенное лекарство переходит из места введения в кровь, которая разносит его по организму и доставляет в различные ткани органов и систем. Этот процесс обозначают термином всасывание (абсорбция). Скорость и полнота всасывания характеризуют биодоступность лекарства, определяют время наступления действия и его силу. Естественно, что при внутривенном и внутриартериальном введении лекарственное вещество «всасывается» сразу и полностью, и его биодоступность составляет 100%.
При всасывании лекарство должно пройти через клеточные мембраны кожи, слизистых оболочек, стенок капилляров, клеточных и субклеточных структур. В зависимости от свойств лекарства и барьеров, через которые оно проникает, а также способа введения все механизмы всасывания можно разделить на четыре основных вида: диффузия (проникновение молекул за счет теплового движения), фильтрация (прохождение молекул через поры под действием давления), активный транспорт (перенос с затратами энергии) и пиноцитоз (захват клеткой макромолекулярных соединений), при котором молекула лекарства как бы продавливается через оболочку мембраны (рисунок 1.2.2). Эти же механизмы транспорта через мембраны используются и при распределении лекарств в организме, и при их выведении. Обратите внимание, что речь идет о тех же процессах, с помощью которых клетка обменивается веществами с окружающей средой.
Некоторые лекарства, принимаемые через рот, всасываются путем простой диффузии в желудке, большинство же из них — в тонком кишечнике, имеющем значительную поверхность (примерно 200 м2) и интенсивное кровоснабжение. Желудок — первая остановка на пути принятых через рот лекарств. Эта остановка довольно короткая. И уже здесь их поджидает первая ловушка: лекарства могут разрушаться при взаимодействии с пищей или пищеварительными соками. Чтобы избежать этого, их помещают в специальные кислотоустойчивые оболочки, растворяющиеся лишь в щелочной среде тонкого кишечника. Задержка в желудке нежелательна, ведь всасывание там происходит сравнительно медленно. Однако есть лекарства, всасывание которых в желудке желательно, поскольку они должны действовать непосредственно на желудок и процесс пищеварения, например, средства, снижающие кислотность желудочного сока путем нейтрализации соляной кислоты (антациды), противоязвенные средства. В желудке происходит также всасывание лекарств, обладающих кислотными свойствами: салициловая кислота, ацетилсалициловая кислота, снотворные средства из группы лекарственных средств, производных барбитуровой кислоты (барбитураты), оказывающие успокаивающее, снотворное, наркозное или противосудорожное действие, и другие.
За счет диффузии всасываются лекарственные вещества и из прямой кишки при ректальномвведении.
Фильтрация через поры мембран встречается значительно реже, так как диаметр этих пор невелик и через них могут пройти только мелкие молекулы.
Наиболее проницаемы для лекарств стенки капилляров, а наименее — кожа, верхний слой которой состоит, в основном, из ороговевших клеток.
Но интенсивность всасывания через кожу может быть увеличена. Вспомним, что питательные кремы и маски наносят на специально подготовленную кожу (удаление избытка ороговевших клеток, очищение пор, улучшение кровоснабжения достигается, например, с помощью водяной бани), а усиления обезболивающего эффекта при воспалении мышц (в медицине это называется миозитом, а в народе говорят — «продуло») добиваются с помощью местного массажа, втирая мази и растворы в больное место.
Всасывание лекарств при сублингвальном применении (под язык) происходит быстрее и интенсивнее, чем из желудочно-кишечного тракта.
Лекарства, принимаемые внутрь (а таких лекарств большинство), всасываются из желудочно-кишечного тракта (желудок, тонкая и толстая кишка), и естественно, что процессы, протекающие в нем, влияют на их всасывание в наибольшей степени.
Конечно, нам было бы очень удобно, если бы все лекарства можно было принимать внутрь. Однако пока этого добиться не удается. Некоторые вещества (например, инсулин) полностью разрушаются ферментами в желудочно-кишечном тракте, а другие (бензилпенициллины) — кислой средой в желудке. Такие лекарства применяют в виде инъекций. Этим же способом пользуются, если необходимо оказать экстренную помощь.
Если лекарство должно оказать действие только на месте введения, его назначают наружно в виде мази, примочек, полоскания и тому подобное. Некоторые препараты, принимаемые в малых дозах (например, нитроглицерин), могут всасываться и через кожу, если их применяют в виде специальных лекарственных форм, например, чрескожных (трансдермальных) терапевтических систем.
Для газообразных и летучих лекарств основным способом является введение в организм с вдыхаемым воздухом (ингаляция). При таком введении всасывание происходит в легких, имеющих обширную поверхность и обильное кровоснабжение. Таким же путем происходит всасывание аэрозолей.
Медицинская практика насчитывает немало примеров ошибочного введения лекарственных форм: известны случаи получения обширных ожогов глаз при закапывании капель, предназначенных для носа или ушей. Ошибочное внутривенное введение растворов для подкожных или внутримышечных инъекций приводило даже к гибели больных. Вот почему нельзя нарушать соответствия между лекарственными формами и путями их введения.
От распределения лекарства в организме зависит скорость наступления фармакологического эффекта, его интенсивность и продолжительность. Ведь для того, чтобы начать действовать, лекарственное вещество должно сконцентрироваться в нужном месте в достаточном количестве и оставаться там определенное время. В большинстве случаев лекарство распределяется в организме неравномерно, в различных тканях его концентрации отличаются в 10 и более раз, хотя в крови, которая питает эти ткани, концентрация его постоянна. Это обусловлено различиями в проницаемости биологических барьеров, интенсивности кровоснабжения тканей и органов.
Кровь разносит лекарство по всему телу, но, если лекарственное вещество прочно соединится с белками крови, то оно так и останется в составе крови, не попадет в другие ткани и не окажет нужного воздействия. Как правило, связывание с белками плазмы крови носит обратимый характер и ведет лишь к увеличению продолжительности действия лекарств.
Клеточные мембраны — главное препятствие на пути молекул лекарственного вещества к месту действия. Различные ткани человека обладают набором мембран с различной пропускной способностью. Легче всего преодолеваются стенки капилляров, самые труднопреодолимые барьеры — между кровью и тканями мозга (гематоэнцефалический барьер или «ворота в мозг») и между кровью матери и плода (плацентарный).
Неравномерность распределения лекарства в организме часто вызывает побочные действия. Рассмотрим следующий пример. Человек заболел воспалением легких (пневмония). Это означает, что у него поражена легочная ткань. Причиной воспаления легких являются микроорганизмы, чаще всего пневмококки. Чтобы с ними справиться, врач назначает, к примеру, сульфадимезин. Масса легочной ткани 1000 г, для воздействия на микробы достаточно 10 мг препарата. Врач, тем не менее, вынужден назначать до 7000 мг сульфадимезина в сутки, так как только при этой дозе обеспечивается нужная концентрация препарата в легких. Оставшаяся часть сульфадимезина накапливается в печени, почках, мышцах и костном мозге, вызывая в них изменения, которые часто осложняют течение болезни и наносят организму серьезный вред. Можно ли уменьшить дозу? Нет, так как в этом случае возбудитель болезни не будет уничтожен.
Есть ли выход? Да. Необходимо научиться управлять распределением лекарств в человеческом организме. Находить лекарственные вещества, способные избирательно накапливаться в определенных тканях. Создавать лекарственные формы, высвобождающие лекарство в тех органах и местах, где необходимо его действие.
И пока эти задачи не будут полностью решены, человечеству не удастся справиться, например, с раком — заболеванием, уносящим многие жизни. Найдены чрезвычайно активные соединения, способные разрушить любую опухолевую ткань. Но … увы! Эти вещества так же активно разрушают и нормальные ткани, а заставить их накапливаться только в тканях опухоли ученые пока не умеют.
В начале главы мы уже говорили о том, что для организма лекарства являются чужеродными веществами, и поэтому он постоянно старается от них избавиться. Для этого организм с помощью ферментов пытается расщепить или связать молекулу лекарственного вещества и, таким образом, облегчить процесс ее выведения из организма. Ферментные системы человека обладают огромной мощностью и позволяют осуществлять в организме процессы, которые в производственных условиях требуют высоких значений температуры, давления и прочее.
Большинство лекарств подвергается превращению в организме — биотрансформации. Лишь небольшое количество препаратов выводится из организма в неизмененном виде. Основными реакциями, которые при этом протекают, являются окисление, восстановление, гидролиз, синтез. В результате этих реакций могут образовываться новые вещества, обладающие более высокой активностью (имизин — дезипрамин), токсичностью (фенацетин — фенетидин) или имеющие собственное фармакологическое действие, отличное от действия принятого лекарства (ипразид — изониазид).
Многие лекарства преобразуются путем присоединения к ним молекул веществ, имеющихся в организме. К последним относятся: глюкуроновая кислота, глицин, метионин, цистеин, уксусная кислота и другие.
Глицин, например, связывает салициловую кислоту и бензойную кислоту, метионин — противотуберкулезное средство этионамид, уксусная кислота соединяется с сульфаниламидными препаратами. Образующиеся продукты, как правило, лишены не только специфической активности, но и, что очень важно — токсичности. Однако при этом возникает другая проблема. Изъятие из оборота важных для нашего организма участников обмена веществ может привести к нарушениям биохимических процессов в целом и, следовательно, отрицательно повлиять на функционирование различных органов и систем. Например, метионин является незаменимой аминокислотой, потребность в ней должна покрываться постоянным поступлением извне. Метионин участвует в реакциях, протекающих при образовании ядерного вещества клеток. Если для обезвреживания лекарства используется слишком много метионина, биохимические процессы нарушаются, и возникают типичные симптомы дефицита этой аминокислоты.
Основную роль в процессе превращения лекарств играют ферменты печени — нашей главной биохимической фабрики по очистке организма от вредных продуктов обмена и всех чужеродных веществ. За счет различных химических реакций сложные нерастворимые молекулы лекарственных веществ расщепляются или переводятся в более легко растворимые формы, что способствует их выведению из организма. При заболеваниях печени (или других состояниях с недостаточной скоростью синтеза или низкой активностью печеночных ферментов) превращение лекарств замедляется, что ведет к увеличению силы и продолжительности их действия.
Активность печеночных ферментов настолько высока, что существует даже такое понятие, как эффект «первого прохождения» через печень. Что же это такое?
Как мы уже знаем, лекарства, которые всасываются из кишечника, разносятся кровью по всему организму только после того, как пройдут через печень, а в этой «химической лаборатории» на них действуют ферменты.
Защитные свойства печени, спасающие нас от ядовитых веществ, становятся мощным и, в некоторых случаях, непреодолимым препятствием для лекарственного вещества. Лишь немногие лекарства способны пройти этот барьер, не потеряв (хотя бы частично) начальной активности.
Эффект «первого прохождения» через печень сильно затрудняет работу лекарства, но печень — естественный защитник организма от чужеродных веществ, поэтому обманывать ее лучше не надо, можно вообще остаться без защиты. Если лекарство быстро (при первом прохождении) разрушается печенью, ищут другие способы введения препаратов. Например, ректально. Известно, что около трети объема крови, движущейся от прямой кишки, минует печень. Это учитывается при создании суппозиториев (или проще говоря — свечей), которые плавятся при температуре человеческого тела и высвобождают лекарство, которое частично (на 1/3) всасывается в общий кровоток, минуя печень. Этот способ введения незаменим также в тех случаях, когда пациент не может глотать или желудок уже не воспринимает никаких лекарств.
Основная часть лекарств после превращения (биотрансформации) или в неизмененном виде выводится из организма с мочой почками. Выведение веществ в этом случае зависит от их растворимости в воде и реакции мочи. Например, при щелочной реакции мочи быстрее выводятся кислотные соединения, а при кислой — щелочные. Эти различия используют и при отравлении (интоксикации) лекарствами, когда, изменяя реакцию мочи приемом соответствующих веществ, добиваются ускоренного выведения из организма этих лекарств (например барбитуратов или алкалоидов). Ускорить выведение лекарств из организма можно и с помощью мочегонных средств при одновременном потреблении большого количества жидкости.
Помимо почечной, в выведении участвуют и другие системы. Некоторые лекарственные вещества (например, тетрациклины, пенициллин, дифенин, колхицин и другие), а также их промежуточные продукты обмена веществ (метаболиты) выводятся с желчью в кишечник, откуда частично удаляются с фекалиями или повторно всасываются в кровь (кишечно-печеночная рециркуляция). Желудочно-кишечным трактом удаляются и те вещества, которые при введении через рот всасываются не полностью.
Газообразные и многие летучие вещества (например, средства для ингаляционного наркоза, небольшая часть принятой дозы алкоголя) выводятся, в основном, легкими. Некоторые препараты выводятся слюнными (йодиды), потовыми, слезными (рифампицин) железами, а также железами желудка (морфин, хинин, никотин) и кишечника (слабые органические кислоты).
Действие лекарства и яда во многом зависит от скорости выведения и возможности ее регулировать. Удерживая лекарство в организме, можно увеличить эффективность лечения, а ускоряя выброс яда, уменьшить последствия отравления.
Медики используют также способность некоторых лекарств накапливаться в тканях и органах на пути выведения и назначают именно то лекарство, которое создает в нужном месте наибольшую концентрацию. Например, при воспалительных заболеваниях мочевыделительной системы применяют вещества, достаточно быстро выводящиеся почками и создающие в них лечебную концентрацию, например, производные нитрофурана (фуразидин, нитрофурантоин и другие). Нецелесообразно при воспалении мочевого пузыря (цистите) лечить пациента тетрациклином или сульфадиметоксином, так как эти средства почками выводятся медленно. В то же время они накапливаются в желчи и могут помочь при воспалительных заболеваниях желчного пузыря и желчных протоков.
С другой стороны, способность лекарств концентрироваться на пути выведения обусловливает в ряде случаев и осложнения при лекарственной терапии. Например, когда в медицине начали использовать сульфаниламиды — вещества с весьма низкой токсичностью, то считали, что никаких осложнений от их применения быть не может. Однако появились сообщения о повреждающем действии сульфаниламидных препаратов на мочевыводящие пути. В почках образовывались камни, известны стали даже случаи смерти от недостаточности почек. В чем же дело? Оказалось, что большинство сульфаниламидов, концентрируясь в мочевыделительной системе, образуют камни в лоханках, мочеточниках, мочевом пузыре. Образовавшиеся камни препятствуют оттоку мочи — отсюда и болевой синдром, и гибель почечной ткани.
Поскольку многие лекарства выводятся почками, становится ясно, почему врачи уменьшают дозы больным с почечной недостаточностью. У таких пациентов лекарства дольше задерживаются в организме и, следовательно, назначение по обычным схемам может привести к передозировке.
Терапевтический лекарственный мониторинг
Статьи
Опубликовано в журнале:
Качественная клиническая практика »» 1 / 2002 А.В. Соколов
Кафедра клинической фармакологии с лабораторией фармакокинетики РГМУ, Москва
Одной из основных задач, стоящих перед клинической фармакокинетикой, является поддержание оптимальной концентрации лекарства в месте действия — оптимизации фармакотерапии. Особенно это касается препаратов, имеющих узкий терапевтический коридор (некоторых антибиотиков, антиаритмиков, циклоспоринов, антиконвульсантов и др.). Эта задача решается с помощью терапевтического лекарственного мониторинга (Therapeutic Drug Monitoring). Для целого ряда лекарственных препаратов назначение так называемых средних доз без учета знания концентрации препарата в крови может приводить к непредсказуемым последствиям. На чем основываются основные принципы проведения терапевтического лекарственного мониторинга (ТЛМ)? После введения препарата в организм его молекулы в месте действия должны находиться в равновесии с молекулами этого препарата в крови. Это означает, что оптимальному терапевтическому эффекту должна соответствовать некая средняя концентрация (или средний диапазон концентраций) препарата в крови пациента. На стадии разработки новых лекарственных препаратов проводятся обязательные фармакокинетические исследования, позволяющие выяснить оптимальный «терапевтический коридор» для многих лекарств, который можно сформулировать как диапаюн концентраций препарата в крови, в пределах которого существует достаточно высокая вероятность получения положительного эффекта и достаточно низкая вероятность появления нежелательных побочных и токсических эффектов.
Врач при назначении лекарства должен решить две основные задачи безопасной фармакотерапии:
- при значительной межиндивидуальный вариации фармакокинетических параметров препарата, приводящей к существенным различиям в конкретных значениях стационарных концен граций в крови пациента (особенно важно внимательно относиться к фармакотерапии у детей, у которых имеются существенные различия в массе тела и скорости метаболизма: нельзя не учитывать и половые различия);
- при нелинейной кинетике препарата нет прямой зависимости междудозой препарата и концентрацией препарата в крови в пределах терапевтического уровня (например, в случае применения фенитоина);
- при очень узком терапевтическом коридоре (опасность получения нежелательных побочных и токсических проявлений);
- при специфическом контингенте пациентов (беременные и кормящие женщины, лица пожилого возраста, грудные дети и т.д.), у которых фармакокинетические параметры, а значит, и границы безопасного терапевтического коридора, значительно отличаются от обычных известных средних значений;
- при нарушениях функции почек, печени или ЖКТ, влияющих на фармакокинетические параметры;
- при политерапии, когда нельзя исключить взаимовлияния нескольких препаратов и трудно смоделировать процессы, приводящие к нормализации фармакокинетических параметров;
- при сомнении в регулярности приема препарата пациентом.
Существует довольно мною различных способов оптимизации фармакотерапии с помощью различного рода номограмм, различных графических методов и применением более современных методов, основанных на использовании современных фармакокинетических компьютерных программ, которым и следует, на наш взгляд, отдавать предпочтение в особо сложных случаях. Важно применение таких программ в случаях нелинейной кинетики препаратапри наличии минимального количества значений концентраций, когда еще не достигнуто стационарное распределение препарата в организме человека.Анализ лекарственных препаратов в биологических пробахОсновой для получения различных фармакокинетических параметров является определение концентраций лекарственного вещества в различных биологических жидкостях (кровь, моча, спинномозговая жидкость, амниотическая жидкость и др.) в определенные моменты времени после приема препарата. Выбор объекта исследования зависит от многих факторов. В первую очередь, он определяется доступностью исследуемого материала. Как правило, наиболее удобными объектами исследования являются кровь и моча. Исследования уровня концентраций лекарств в крови — наиболее распространенный фармакокинетическии подход для оптимизации фармакотерапии еще и потому, что фармакокинетические параметры, полученные в этом случае, можно коррелировать с данными биохимического анализа крови пациента. Существует большое число самых различных методов определения концентраций лекарств в биологических жидкостях: xроматографические, микробиологические, спектрофотометрические, полярографические, иммунологические (радиоиммунные, иммуноэнзимные), радиоизотопные и другге методы, основанные на различных физико-химических свойствах исследуемых материалов. Все вышеназванные методы достаточно хорошо описаны в мировой литературе и довольно широко применяются при проведении ТЛМ. Безусловно, каждый метод определения концентрации обладает определенными достоинствами и недостатками. Требования, предъявляемыми к методу в нескольких словах, следующие: чувствительность определения, экспрессность анализа, точность анализа, возможность работы с малым объемом биоматериала, стоимость анализа. Иммунологические методы, к безусловным достоинствам которых можно отнести относительную простоту и экспрессность проведения непосредственных измерений, занимают по праву одно из ведущих мест. Однако необходимо знать, что применение подобных методов сопряжено с определенными трудностями: необходимы специальная аппаратура и наборы, существующие далеко не для всех лекарственных средств, для которых ТЛМ остро необходим. Кроме этого, стоимость наборов достаточно высока и определение, даже разовое, обходится достаточно дорого. Далее, как правило, все наборы выпускаются различными зарубежными фирмами и, раз познав всю простоту и легкость решения проблемы, вы становитесь «заложником» фирмы-производителя. Применение микробиологических методов в нашей стране, как правило, ограничивается исследованием достаточно широкого диапазона антибиотических средств. Но, следует учитывать, что ни о какой экспрессности, чувствительности и точности при использовании данного метода речь идти не может. Применение таких методов, как полярографические, различные фотометрические методы при проведении ТЛМ нецелесообразно из-за высокой специфичности данных методов, недостаточно высокой чувствительности определения, связанной с целым рядом факторов и достаточно малой универсальностью применения. Гораздо более перспективно в существующих условиях использовать различные хроматографические методы анализа лекарств. Преимущества данных методов налицо — высокая чувствительность и точность определения, огромная универсальность применения (с помощью хроматографических методов возможно исследование более 95% всехлекарственных средств, используемых в современной фармакотерапии), достаточно большая экспрессность, возможность быстрого переключения с определения одного препарата на другой (при условии наличия метода определения), относительно невысокая стоимость анализа (заключающаяся в малых количествах используемых реактивов и прочих затратах). Конечно, при применении хроматографических методов необходимо иметь аппаратуру, которая имеет достаточно высокую стоимость и высококвалифицированных специалистов анализа. Однако при проведении массовых анализов эти затраты, как правило, довольно быстро окупаются. Когда-то выдающийся французский ученый XIX века Клод Бернар сказал: «Создание хорошего метода приносит иногда науке больше пользы, чем высокие теоретические соображения». Яркой иллюстрацией этой мысли является появление хроматографии. Рассмотрим основные принципы проведения хроматографического анализа. В 1903 г. русский ученый М.С. Цвет, изучая хлорофилл, должен был найти доказательства выдвинутого им предположения о сложности состава хлорофилла, который до этого считался индивидуальным веществом. В 1906 г. на собрании Московского общества естествоиспытателей М.С. Цвет сделал доклад о своей работе, в результате которой были выделены составляющие хлорофилл-компоненты. Одновременно М.С. Цвет изложил разработанный им новый метод разделения сложных веществ. Со времени открытия метода хроматографического анализа принципы, заложенные в основу этого метода разделения сложных смесей химических веществ, по сути не изменился. В основе его лежит принцип различия в сорбционной способности каждого химического вещества на том или ином сорбенте (веществе с большой адсорбционной емкостью). Продвигаемая носителем (элюентом) вдоль сорбента смесь веществ из-за разной величины адсорбционных свойств в одних и тех же условиях подвергается разделению (подобно разделению по температурам кипения, которое происходя при перегонке). При этом слой сорбента может находиться в виде тонкого слоя на пластинке, а продвигать смесь веществ может растворитель (или смесь растворителей) за счет капиллярных сил. В этом случае данный метод разделения будет называться тонкослойной хроматографией (ТСХ), сорбент может быть упакован в достаточно тонкую стеклянную или металлическую трубку, а анализируемая смесь продвигается вдоль колонки либо газом (газноситель) и тогда метод называется «газовая хроматография» (ГХ), либо потоком ра створителя (смесью растворителей), подаваемого в колонку под давлением (иногда довольно значительным) с помощью насоса — в этом случае мы имеем дело с жидкостной хроматографией (ВЭЖХ). Проведя хроматографическое разделение, необходимо обработать полученные данные, определить, что мы получили в результате анализа и в каком количестве, т.е. провести качественный и количественный анализы. Такие анализы проводятся обычно с помощью приборов, называемых детекторами, а запись полученного сигна ла осуществляется с помощью электронных интеграторов, самописцев или современных компьютерных программ в виде аналоговой формы электронного сигнала детектора. В результате мы получаем хроматограмму — картину разделения, где каждому разделенному веществу соответствует индивидуальный пик. Для проведения количественного определения полученные пики обсчитываются, калибруются и с помощью различных электронных устройств оператор получает полную количественную картину состава исследованного образца. Таким примерно образом проводится обычный хроматографический анализ многокомпонентных смесей. Для успешного решения задач количественного анализа хроматографическими методами необходимо проводить достаточно сложные процедуры подготовки проб для анализа. Вопросы подготовки проб, выбора метода анализа, разработки метода, количественной обработки полученных данных требуют серьезного отношения и использования профессионально подготовленных специалистов. Собственно весь процесс проведения любых фармакокинетических исследований предполагает использование специалистов самого различного уровня -врачей, биохимиков, химиков-аналитиков, математиков. От тщательности проведения каждого отдельного этапа зависит результат всей работы. Большинство фармакокинетических методов исследования базируется на изучении уровня концентрации того или иного препарата в крови пациента. При возникновении той или иной фармакокинетической задачи перед исследователем возникает вопрос: каким методом предстоит воспользоваться — ГЖХ, ТСХ, ВЭЖХ, ГХ-МС, иммунным, радиологическим, полярографическим или каким-либо другим. Большое внимание следует уделять вопросам подготовки и обработки биологических проб для анализа. От того, какой биологический объект будет изучаться, зависит как выбор метода анализа, так и способ его проведения. Оттого, каким методом предстоит воспользоваться, зависит многое, например способ подготовки проб для анализа. В основном процесс подготовки проб для анализа сводится к следующим процедурам: 1. Получение сыворотки крови. 2. Извлечение препарата в удобной для анализа форме (одно- или многократная экстракция препарата химическими растворителями или процесс осаждения белков тем или иным реагентом. В последнем случае необходимо помнить, что при осаждении белков сыворотки крови происходит разбавление образца, что влияет на чувствительность метода анализа). 3. Сохранение образца в виде сывороточных экстрактов, сыворотки крови, упаренных образцов в сухом виде и т.д. для последующего хроматографического анализа. Следует помнить, что при проведении фармакокинетических исследований некоторые препараты довольно быстро подвергаются биотрансформации (окислению, гидролизу и т.п.). Исходя из этого, каждый исследователь определяет минимальные и максимальные сроки проведения анализа. В процессе разработки метода следует также рассмотреть и исключить (или по возможности стандартизироиать} все возможные варианты потерь вещества при проведении основных и вспомогательных операций, чтобы избежать ошибок в анализе. Как же осуществляется выбор метода анализа лекарственного препарата? Очень часто этот выбор зависит от наличия той или иной аппаратуры в распоряжении исследователя. Требования, предъявляемые к выбранному методу анализа, можно сформулировать следующим образом. Методанализадолжен бытьдостаточно чувствительным для получения надежных результатов, воспроизводимым, дешевым и достаточно экспрессным. Процесс подготовки проб для анализа недолжен занимать много времени и должен быть достаточно простым и воспроизводимым. Выбранный методдолжен обладать большой производительностью. Примерно такие же процедуры необходимы при проведении газохроматографического анализа, с той лишь разницей, что при данном анализе практически исключена возможность анализа водосодержащих проб. Кроме того, при проведении подобного анализа возникает необходимость применения высокочистых сжатых газов (азот, гелий, водород и воздух), что соответственно удорожает весь анализ. Итак, мы знаем пути получения точных данных по уровню концентрации лекарственного препарата в той или иной биологической среде. Эти данные необходимо использовать при проведении фармакокинетических исследований в клинических условиях. Крайне важной областью применения этих данных и является терапевтический лекарственный мониторинг.Основные понятия фармакокинетикиИнтенсивность эффекта применяемого препарата находится в прямой зависимости с его концентрацией в месте действия. К сожалению, далеко не всегда возможно определение концентрации препарата в месте действия. Однако если принять предположение, что концентрация препарата в месте действия находится в зависимости от его уровня в каком-либо другом объекте, задача расчета достижения оптимальной концентрации препарата в месте действия представляется вполне достижимой. Описать все детали процесса распределения препарата в организме, во всех органах и тканях практически невозможно, или, по кранней мере, очень сложно. Однако это не всегда является необходимым. Во многих случаях бывает достаточно чисто формально представить организм в виде одной или более камер (компартментов), определить связи между этими камерами и составить материальный баланс. При этом появляется возможность составить достаточно упрощенные математические модели кинетики изучаемого препарата, в состав которых входит лишь ограниченное число измеряемых параметров. Такой упрощенный подход к математическому моделированию позволяет составлять довольно точные математические прогнозы получения желаемого уровня концентрации препарата в месте действия, основываясь на использовании хорошо разработанного математического аппарата и теории оптимизации. Такое моделирование называется камерным, или компартментным. При этом камера — это та часть организма, в которой препарат распределен равномерно. Для целей фармакокинетического анализа выбирается такое минимальное число камер, которое в данном случае необходимо для составления модели по имеющимся ограниченным опытным данным. Минимальным количеством камер, как это можно предположить, является единица. Математическая модель при этом будет однокамерной. При однокамерной модели предполагается некая фармакокинетическая однородность всех тканей, в которые проник препарат. Иными словами, предполагается, что сразу после введения препарата его концентрация в этой единственной камере становится равновесной и убывает по моноэкспоненциальному закону без видимой фазы распределения. В соответствии с законамилинейной кинетики скорость изменения количества препарата в этой единственной камере пропорциональна его количеству в камере. Для того чтобы перейти от количества препарата в камере к его концентрации, вводят коэффициент пропорциональности, который называют кажущийся объем распределения препарата (не эквивалентен физиологическому объему тканей камеры). Объем распределения препарата (Vd) является одним из наиболее важных фармакокинетических параметров и обычно рассчитывается на массу тела (л/кг). Величина объема распределения в рамках однокамерной модели при внутривенном введении равна такому условному объему жидкости, в котором нужно растворить всю попавшую в организм дозу препарата, чтобы получилась концентрация, равная начальной концентрации в крови (Сo). При различных способах введения препарата структура однокамерной модели может быть представлена двумя различными вариантами. Внутривенное введение или постоянная инфузия (рис. А) или постепенное введение препарата в камеру (рис. Б) из некоторого депо (при этом учитывается процесс абсорбции при внесосудистом введении).
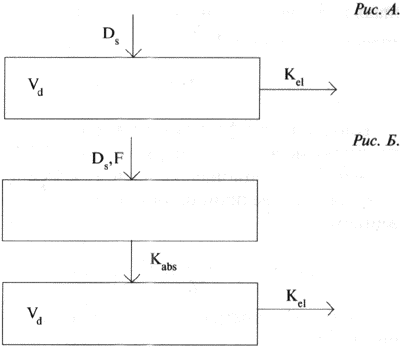
При поступлении препарата в системный кровоток из депо его концентрация постепенно возрастает, достигая максимального значения Сmax в момент времени Тmax и затем начинает убывать. Если считается, что процесс абсорбции имеет линейный характер (скорость процесса прямо пропорциональна количеству или концентрации препарата в системе), то скорость этого процесса характеризуется константой скоростью абсорбции k(abs) измеряемой в л/ч. В случае линейного процесса эту константу можно выразить через период полувсасывания Т1/2(abs) — время, за которое всасывается половина введенной дозы препарата: k(abs) = ln2 / Т1/2(abs) При внесосудистом введении лекарства не все количество препарата попадает в системный кровоток, поэтому в фармакокинетике пользуются термином биодоступность — F. Биодоступность характеризует ту часть введенной дозы препарата, которая достигает системного кровотока. Определяется биодоступность при сравнении площадей под кривой «концентрация — время» — AUC для исследуемого препарата и препарата сравнения: F = AUC(test) / AUC(standart) Биодоступность бывает относительная и абсолютная. Когда в качестве стандарта используют данные о внутривенном введении этого же препарата, а в качестве тестового препарата — данные о внесосудистом введении, то речь идет об абсолютной биодоступности. Если же используются данные о внесосудистом введении различных лекарственных форм одного и того же препарата, например изготовленных разными фирмами, то речь идет об оценке относительной биодоступности. При этом в качестве стандарта выбирается хорошо известный лекарственный препарат. В рамках однокамерной модели при внутривенном введении площадь под кривой «концентрация — время» выражается следующим образом: AUC = Co / K(el) Площадь под фармакокинетической кривой пропорциональна дозе препарата, попавшей в организм, обратно пропорциональна общему клиренсу препарата и связана с величиной объема распределения выражением: Vd = D / (K(el) * AUC) где D — однократная доза препарата, введенная внутривенно. Очень важно отметить, что параллельно с процессом абсорбции происходит процесс выведения препарата — элиминация. Элиминация препарата, как правило, складывается из двух процессов — биотрансформации (или метаболизма) и экскреции препарата. Метаболическое превращение препарата происходит главным образом в печени (за счет работы ферментов, которые гидролизуют, окисляют, восстанавливают, алкилируют, ацетилируют и т.п. лекарство). Часто продукт метаболической деятельности организма обладает большей активностью, чем введенное лекарство (новокаинамид, эналаприлат и др.). При линейном характере процесса выведения в рамках однокамерной модели элиминация препарата описывается моноэкспоненциальной зависимостью и характеризуется константой скоростью элиминации K(el) (л/ч). Этот параметр в общем виде представляет собой сумму скоростей выведения почечным и непочечным путями. Для однократного внутривенного введения дозы препарата D уменьшение концентрации препарата в единственной камере происходя в соответствии с уравнением: C(t) = (D / Vd) — exp(-K(el) * t) Для такой модели может использоваться и другой показатель, характеризующий процесс выведения препарата — период полуэлиминации, всем хорошо известный параматр Т1/2, который связан с константой скорости элиминации выражением: Т1/2 = 0,693 / K(el) Количественную оценку элиминируемого препарата дает клиренс (Сl) — объем крови, очищаемый от препарата в единицу времени (мл/ч или л/ч). Для препаратов, которые быстро распределяются и чье поведение может быть описано в рамках линейной модели, клиренс определяется выражением: Сl = Vd * K(el) При линейном характере процессов абсорбции и элиминации изменение концентрации препарата во времени после однократного внесосудистого приема описывается уравнением: C(t) = (K(abs) * F * D) / [Vd * (K(abs) — K(el))] — [exp(-K(el) * t) — exp(-K(abs) * t)] Таким образом, при определенных значениях биодоступности препарата (F) и получаемой пациентом дозы (D) форма фармакокинетической кривой определяется соотношением основных параметров кинетики — Vd, K(abs) и K(el) Если препарат вводится в постоянной дозе через фиксированные интервалы времени, меньшие, чем время элиминации препарата, то его концентрация в крови возрастает ступенчатым образом, а затем наступает период, когда в каждом интервале между приемом очередных доз препарата количество всасывающегося препарата равно количеству элиминируемого. Это состояние называется стационарным или Steady stale, а концентрация, достигнутая при этом, называется стационарной и обозначается Сss. При одной и той же скорости абсорбции чем длиннее период полуэлиминации или полувыведения препарата по отношению к интервалу дозирования, тем медленнее достигается стационарный уровень. Иными словами, чем больше T1/2 выведения препарата, тем больше времени потребуется для достижения стационарного уровня концентрации. На практике принято считать, что состояние равновесия достигается по прошествии 4-5 периодов полувыведения препарата. Для выбора оптимального интервала дозирования препарата очень важна информация о ширине терапевтического коридора (или о максимально допустимой амплитуде колебаний концентрации препарата в крови) и о временах полувыведения. Целью врача является удерживать концентрацию препарата в рамках этого коридора, подбирая оптимальные интервалы дозирования (т). Изменение концентрации препарата во времени при многократном введении одинаковых доз (D) через равные промежутки времени (т) в стационарном режиме описывается уравнением: Сss(t) = (K(abs) * F * D) / [Vd * (K(abs) — K(el))] * [exp(-K(el)t) / 1 — exp(K(el)т) — exp(-K(abs)t) / 1 — exp(K(abs)т)] В том случае, если определенную концентрацию в крови необходимо создать немедленно и затем поддерживать ее на этом уровне, дается нагрузочная доза препарата, а затем — поддерживающие. В тех случаях, когда с помощью простейших однокамерных моделей на основе экспериментально полученных данных не удается приемлемо описать процессы, применяются двухкамерные (и более) модели. При этом считается, что в первой камере происходит быстрое распределение препарата и затем более медленный обмен с другой (или другими) периферической камерой. В таких случаях так же, как и при однокамерных моделях, строятся дифференциальные уравнения материального баланса. Эти многокамерные модели с помощью таких уравнений могут описывать различные варианты введения препарата. Например, двухкамерная модель может быть схематично изображена в виде центральной и периферической камер, отличающихся скоростями взаимного проникновения препарата (Рис. В).
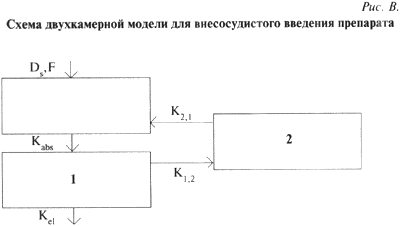
При использовании таких моделей появляются дополнительные параметры — константы скоростей обмена между камерами (К(2,1) и К(1,2)). Теперь, зная фармакокинетические параметры, могут быть составлены уравнения для двух и более камер и соответственно построены фармакокинетические кривые для этих камер. При приеме препарата через определенные, равные между собой промежутки времени в крови вскоре появляется так называемый плато-эффект, т.е. устанавливается постоянный уровень препарата. Величину этого уровня можно оценить по приближенному уравнению:Ccp ~ (1,5 * D * F* t1/2) / (Vd * т) где F — доля препарата, всосавшаяся в организм при пероральном приеме (меняется от 0 до 1 ): т — интервал между дозированием лекарства. Для оценки скорости изменения концентрации препарата в крови в процессе лечения можно рассчитать число повторных приемов, которые создадут концентрацию, отличающуюся от стационарной не более чем на 10%. Подобные расчеты возможны при условии, что в течение курса лечения величина k(эл) существенно не изменяется (препараты, для которых не отмечены явления индукции или угнетения метаболизирующих ферментов). Таким образом, мы убедились, что фармакокинетические методы позволяют произвести расчет оптимальной и индивидуальной дозировки препарата. При этом должны выполняться следующие условия: 1. Концентрация препарата в крови должна достигнуть желаемого стабильного уровня сразу после первичного приема (Сss°). 2. Минимальная концентрация препарата в крови, т.е. концентрация перед очередным приемом, недолжна быть ниже эффективной терапевтической концентрации (Сэф). 3. Максимальная концентрация препарата не больше безопасной концентрации (Сs). Для выполнения этих условий можно вывести следующие уравнения для расчета начальной дозы Dн и поддерживающих доз Dп: Dн = Сэф * Vd * ek(эл)*т / f Dп = Сэф * Vd * (ek(эл)*т — 1) / f Для безопасного лечения рассчитанными дозами интервалы между приемами препарата (t) должны быть не больше максимального интервала (tmax): т(max) = 3,33 * t1/2* lg[1 + Y(S-1)] где Y — специальный безразмерный параметр, S — характеристика терапевтической широты препарата S = Cs / Сэф. Величина Y рассчитывается по специальным таблицам или экспериментальным данным. Однако подобные расчеты можно с достаточной надежностью вести и по уравнению: т(max) = 1,3 + S * (0,24 + 0,54 * t1/2) Эти зависимость вполне логична — чем медленнее выводится препарат (чем больше t1/2), тем реже его дозировка. Учитывая, что значения т(max) являются по определению максимально допустимыми интервалами между дозированием препарата, при расчете начальных и поддерживающих дозировок следует пользоваться меньшими значениями t, при этом достаточно хорошие результаты достигаются при использовании величины т, равной примерно половине т(max). Все, что мы рассмотрели в смысле фармакокинетического моделирования, основано на предположении о линейной кинетике, которое может быть сформулировано следующим образом:
- фармакокинетические параметры не зависят от дозы препарата,
- площадь под фармакокинетической кривой пропорциональна дозе при любом способе введения,
- количество препарата в камерах также пропорционально дозе препарата.
В случае невыполнения хотя бы одного из этих условий фармакокинетические данные необходимо описывать нелинейными моделями. Одной из наиболее распространенных нелинейных фармакокинетических моделей является однокамерная (или двухкамерная) модель, элиминация препарата из центральной камеры которой происходит в результате метаболизма в соответствии с законом Михаэлиса-Ментен: dC/dt = -Vmax * С(t) / [Кm + С(t)], где С(t) — концентрация препарата, Vmax — максимальный объем продуктов метаболита, Кm — константа Михаэлиса (параметры Михаэлиса-Ментен). При этом, если процесс метаболизма далек от насыщения, кинетика выведения становится аналогичной линейной однокамерной модели со скоростью выведения: Vmax / Кm. Если метаболизм носит насыщенный характер (высокие концентрации препарата в крови), процесс описывается кинетикой нулевого порядка: dC/dt = -Vmax Следует учитывать, что в некоторых случаях большую роль играет активность метаболитов. Так, при анализе связи между антиаритмическим эффектом новокаинамида и его стационарными уровнями необходимо обязательно учитывать присутствие в крови его активного метаболита N-ацетилновокаинамида, который выводится из организма в 2-3 раза медленнее самою препарата. Незнание этого может привести к интоксикации. Большое значение имеют методы определения концентрации препарата в крови, о которых мы уже говорили. При спектрофлюорометрическом определении хинидина определяется суммарный уровень как самого препарата, так и его малоактивных метаболитов, что приводит к искажению результатов, давая завышенные почти вдвое значения концентраций. Для значительного числа лекарственных препаратов обнаруживается достаточно высокая линейная корреляция между величиной наблюдаемого фармакологического эффекта и уровнем препарата в крови, т.е. имеет место зависимость: Е = а * Сp + b где E — терапевтический эффект, Сp концентрация препарата в крови. Например, антиаритмический эффект новокаинамида хорошо коррелирует с концентрацией препарата в крови (коэффициент корреляции r=0,81-0,92). При исследовании бронхорасширяющего действия теофиллина у больных с астмой эффект выражался в изменении объема легких при максимальном выдохе. Для анализа связи эффекта с концентрацией теофиллина в крови было выведено уравнение: Е = 63% * Сp / (10 + Сp) Это выражение позволяя сделать несколько важных для клиники выводов:
- даже при максимально возможном эффекте теофиллин у больных астмой не сможет полностью нормализовать функцию органов дыхания в связи с необратимой обструкцией дыхательных путей,
- создание в крови уровня теофиллина, равного 10 мкг/мл, приведет к улучшению исходного объема лекгих (31,5% нормального уровня, при этом обычно наблюдается степень бронходнлатации, достаточная для снятия астматического приступа), в то же время увеличение концентрации теофиллина вдвое приведет к незначительному увеличению эффекта, но создаст опасность интоксикации.
Известны два принципиально различных типа ситуаций, в которых проведение ТЛМ может оказаться достаточно полезным. 1. Терапия проводилась и течение достаточно длительного времени и уже имеются клинические данные о реакции пациента на проводимую терапию. В этом случае ТЛМ может помочь избежать возможных побочных эффектов, а в случае неэффективности проводимой терапии ТЛМ ответить на вопрос о получении лучшего эффекта при изменении режима дозирования. 2. Выбор начального режимадозирования, когда реакция пациента на назначаемый препарат еще не известна. Основная задача здесь — прогнозирование индивидуальной кинетики на основе априорной информации лишь о фармакокинетике данного препарата у «среднего» пациента. При этом учитываются масса тела, рост, пол, возраст, почечная функция данного пациента. Таким образом стараются подобрать начальный режим дозирования, чтобы прогнозируемая концентрация держалась в пределах терапевтического коридора. В обоих случаях следует отметить, что основная цель — создание уровня препарата в крови, которого вы хотели бы достичь в ходе данной терапии. Выбор этого оптимального уровня в любом случае остается за лечащим врачом, а фармакокинетический подход, программное обеспечение лишь помогают рассчитать наилучший режим для достижения поставленной врачом цели. По мнению многих авторов, существует два абсолютных показания для проведения ТЛМ антиконвульсантов: отсутствие положительного эффекта при регулярном получении пациентом средних и высоких доз препарата и клинических проявлениях побочных эффектов. По нашему мнению, ТЛМ может оказаться полезным в следующих ситуациях:
- при первом визите к врачу при получении ранее антиконвульсантной терапии для определения создавшегося уровня препарата в крови;
- для определения стационарной концентрации после назначения препарата или корректировке терапии;
- для определения стационарной концентрации после назначения или отмены других препаратов при комбинированной терапии;
- рутинное исследование 1-2 раза в год после достижения положительных результатов от проводимой терапии (чаще для детей, получающих фенитоин из-за риска аккумуляции препарата);
- при беременности;
- при решении вопроса о прерывании терапии
При проведении ТЛМ антиконвульсантов для обоснования его необходимости мы основывались на наличии или отсутствии корреляционной связи между дозой препарата и его концентрацией в крови у различных пациентов и всей популяции в целом. Если при исследовании выявляется слабая связь или, напротив, достоверной корреляционной связи нет вообще, это означает, что на основе средних фармакокинетических параметров невозможно составить правильны и прогноз по уровню препарата по вводимой дозе. В таком случае необходима индивидуализация дозирования на основе мониторинга. Исследование реакций конкретного пациента на различные дозы препарата стало возможно потому, что наши популяции включали различных пациентов, уровни препаратов в крови которых определялись в различные периоды терапии при различных дозах. Это означает возможность прогноза (с достаточной точностью) уровня препарата в крови при получении пациентом других дозировок. Для проверки работоспособности наших прогнозов уровня исследуемого препарата мы использовали, как правило, данные двух значений концентрации (перед очередным приемом препарата и через несколько часов после приема). По результатам анализа сделанных прогнозов можно сделать вывод, что изменение уровня определяемых нами антиконвульсантов в крови конкретного пациента при изменении терапии может быть достаточно хорошо предсказано даже на основании всего двух измерений концентрации препарата. ТЛМ антиконвульсантов будет давать положительный эффект лишь в том случае, если он проводится грамотно (это предполагает качественное определение концентраций препаратов в крови, правильный выбор моментов взятия образцов крови, применение адекватных методов расчета, тщательное ведение документации). Документироваться должна следующая информация:
- проводимая терапия до начала мониторинга;
- схема дозирования при любых назначениях или отмене препарата;
- уровни концентрации препарата;
- частота и тяжесть приступов при различных схемах дозирования;
- наличие побочных эффектов.
Оптимизация фармакотерапии должна проводиться на основе анализа всей этой конкретной информации, а не только на основании формальных расчетов, исходя из 1-2 измерений уровня препарата в крови. Как уже указывалось, огромное значение ТЛМ приобретает при лечении эпилепсии. Нами разработаны методы определения практически всех антиконвульсантов в крови человека, которые применяются при терапии данного заболевания (карбамазепин, дифенин, суксилеп, фенобарбитал и его производные, вальпроевая кислота и вальпроаты). Методы определения препаратов основаны на применении ВЭЖХ. Разработанные методы позволяют оперативно и с большой точностью проводить анализы (полное время одного анализа на содержание антиконвульсантов не превышает 1,5 ч), что, в свою очередь, дает возможность заниматься оперативной коррекцией дозировки препаратов, выбирать необходимые интервалы дозирования, одним словом — оптимизировать фармикотерапию данного заболевания. С помощью методов популяционного моделирования мы проанализировали результаты мониторирования основных антиэпилептических средств при монотерапии, а также некоторых их сочетаний при политерапии для различных возрастных групп больных эпилепсией. Изучалась фармакокинетика таких широко применяемых препаратов, как карбамазепин, фенобарбитал, фенитоин и производные вальпроевой кислоты — вальпроаты (депакин). По возрастным признакам наши популяции были сформированы из детей и взрослых пациентов. Все пациенты постоянно получали антиконвульсанты и, с точки зрения фармакокинетики, находились в «стационарном» состоянии.Карбамазепин (монотерапия)
| Дети | Взрослые | |
| Количество пациентов | 90 | 99 |
| Возраст, лет | 9,6+/-6,4 | 27,4+/-11,9 |
| Дозы, мг/кг/сут | 16,5+/-9,6 | 9,2+/-5,0 |
Bальпроаты (монотерапия)
| Дети | Взрослые | |
| Количество пациентов | 53 | 37 |
| Возраст, лет | 8,3+/-3,9 | 24,3+/-9,7 |
| Дозы, мг/кг/сут | 34,5+/-17,8 | 17,6+/-6,5 |
Фенобарбитал (монотерапия)
| Дети | Взрослые | |
| Количество пациентов | 28 | 36 |
| Возраст, лет | 11,7+/-3,1 | 26,9+/-8,2 |
| Дозы, мг/кг/сут | 4,5+/-2,8 | 2,6+/-1,7 |
По данным медстатистики Центральной детской республиканской больницы, полученным на основании наших результатов (1992), существенно сокращается время нахождения пациента в стационаре (вместо обычных 20-45, а в некоторых сложных случаях и до 60 дней процесс подбора оптимальной терапии с достаточно высоким положительным эффектом длится не более 12-15 дней), что существенно сокращает затраты на лечение. Такие данные получены на достаточно представительном контингенте больных — более 500 человек. Проведено фармакокинетическое исследование карбамазепина и некоторыхдругихангиконвульсантов на основе терапевтического лекарственного мониторинга (ТЛМ) с применением высокоэффективной жидкостной хроматографии у детей с судорожным синдромом. Все исследуемые пациенты были разделены на группы, которые были рандомизированы по полу и возрасту Полученные результаты дают основание считать, что в возрастной группе 9-14 лет некоторые фармакокинетические параметры существенно различаются у мальчиков и девочек. Так, отмечено достоверное снижение клиренса кабамазепина у девочек (почти в два раза) по сравнению с мальчиками той же возрастной группы, что влечет за собой двукратное уменьшение дозировки препарата. Однако после 14 лет половые признаки перестают оказывать влияние на фармакокинетику препарата. Осуществление контроля за сывороточными концентрациями антиконвульсантов особенно важно при их совместном применении, так как в этом случае довольно часто происходит изменение элиминации какого-либо из антиконвульсантов. ТЛМ противосудорожных препаратов обычно проводится на 3-5-й день после начала лечения. На основании полученных данных с использованием специальных компьютерных программ проводится определение оптимальной дозы препарата, а через неделю осуществляется контрольное определение сывороточной концентрации с целью коррекции дозировки. Таким образом, используя данные ТЛМ, возраст и пол больного, а также эффективность проводимой терапии, можно рассчитать оптимальный режим дозирования антиконвульсантов. По данным двух неврологических отделений РДКБ, в которых проводилось данное исследование, больные, получавшие противосудорожные препараты, находились в стационаре для подбора противосудорожной терапии 10-16 дней (средние данные) вместо обычных 2160 дней. При этом уменьшилось число случаев резистентности, неясных побочных реакций и общее число токсических проявлений или неполного, нестабильного клинического эффекта в связи с ранним выявлением случаев истинной и приобретенной в результате длительного приема одного препарата резистентности. Многим амбулаторным больным с помощью ТЛМ удалось оптимизировать дозировку антиконвульсантов без госпитализации, что ранее было практически невозможно. Приведем несколько примеров практического использования данных ТЛМ в клинических условиях. Так, на базе 1-го неврологического отделения ЦДРКБ на 54 пациентах нами было проведено фармакокинетическое исследование карбамазепина на основе ТЛМ с применением ВЭЖХ у детей с судорожным синдромом. Все дети получали практически одну начальную дозу карбамазепина — 22-25 мг/кг/сут с интервалом дозирования 8 ч. Пробы крови для определения концентрации препарата отбирались из локтевой вены путем венопункции перед очередным приемом препарата и через 1,5 ч после приема. Исследование проводилось через 5 дней после начала лечения. После определения концентрации препарата в крови оказалось, что всех больных можно разделить на четыре условные группы. В первой группе (наиболее многочисленная — 25 человек) концентрация карбамазепина (как минимальная, так и максимальная) находилась в оптимальной области терапевтического диапазона (определяемого для этого препарата 4-10 мкг/ мл). В этой группе была достигнута высокая эффективность лечения без наблюдаемых побочных эффектов. Во второй группе (17 человек), в которой уровень минимально и максимальной концентрации карбамазепина лежал выше терапевтического коридора, отмечались типичные побочные эффекты, связанные с передозировкой препарата. В третьей группе (5 человек) минимальная концентрация препарата находилась ниже терапевтического диапазона, а максимальная концентрация лежала в нижней части диапазона. В этой группе в большинстве случаев наблюдался или незначительный терапевтический эффект или его не было вовсе. И, наконец, в четвертой группе, состоявшей из 7 человек, минимальная концентрация препарата лежала в верхней части терапевтического диапазона, а максимальная находилась значительно выше, с соответствующими для подобных случаев эффектами повторяющихся симптомов легкой передозировки препарата при наличии терапевтического эффекта. После проведения расчетов оптимальной дозы выявилось практически полное равенство между расчетной и реальной дозами в первой группе пациентов, необходимость существенного (примерно на 40%) снижения дозы во второй группе больных, корректировки дозы в сторону некоторого увеличения в третьей группе и некоторого уменьшен ия дозировки с соответствующими изменениями интервалов приема препарата в четвертой группе. Нами также было проведено несколько отличающееся от предыдущего исследование фармакокинетики карбамазепина на основе проведения ТЛ М у достаточно представительной группы детей (42 человека) с судорожным синдромом. Полученные в этом исследовании результаты дают основание считать, что в возрастной группе 9-15 лет некоторые фармакокинетические параметры имеют существенные половые отличия. Так, отмечено вполне достоверное (примерно на 45%) снижение клиренса препарата у девочек по сравнению с мальчиками той же возрастной группы, что влечет за собой практически двукратное уменьшение дозировки препарата. Однако по достижении 14-15 лет половые признаки перестают оказывать столь очевидное влияние на фармакокинетику препарата. Приведем один пример успешного применения популяционного моделирования для оптимизации фармакотерапии у пациентки С. в возрасте 16 лет, масса — 57 кг, рост — 162 см, которая находилась на монотерапии карбамазепином. Пациентка принимала карбамазепин по следующей схеме: утро — 400 мг, днем 300 мг и вечером — 400 мг в течение 4 лет. По данным мониторирования (Сmin — 7,8 мкг/мл, Сmax — 12,7 мкг/мл) явлений передозировки не было, однако больную беспокоили регулярные судорожные приступы перед пробуждением. Измеренные концентрации препарата в крови говорили о практической близости к верхней потенциально опасной границе терапевтического диапазона Сmin — 9,1 мкг/мл, Сmax — 13,0 мкг/мл. С помощью популяционного подхода установлено, что прогнозируемый минимум ниже 9 мкг/мл возможен именно во время наблюдения приступов. По нашим расчетам, для купирования приступов у больной перед пробуждением должна сохраняться концентрация карбамазепина (в 6-7 часов утра), равная 8,2 мкг/мл. Исходя из проведенных расчетов, было два возможных варианта изменения терапии: 1-й вариант — увеличить вечернюю дозу на 100 мг и тогда уровень препарата не будет опускаться ниже 9 мкг/мл (Сmin — 9,0 мкг/мл, Сmax — 13,8 мкг/мл), но амплитуда разброса между максимальным и минимальным значением существенно возрастет. По второму варианту дозу дневного приема препарата необходимо увеличить и принимать по 400 мг ровно через 8 часов. Тогда ожидаемый прогноз по минимальному уровню препарата в крови (Сmin — 9,3 мкг/ мл, Сmax — 13,3 мкг/мл) будет таким же, как и в первом случае, но разброс окажется существенно меньше, а значит, и риск передозировки будет ниже. Нами была выбрана вторая схема, так как разница между минимальной и максимальной концентрацией препарата в этом случае предполагалась несколько меньше, при этом реальные измеренные концентрации карбамазепина составили: Сmin — 9,2 мкг/мл, Сmax — 13,1 мкг/мл. На основании результатов ТЛМ карбамазепина у беременных женщин 20-30 лет с характерным эписиндромом нами совместно с сотрудниками кафедры нейрохирургии и неврологии ММСИ под руководством чл.-корр. АМН, профессора В.А. Карпова установлена достоверная связь между суточной дозой карбамазепина и концентрацией женских половых гормонов. Так, при суточной дозе свыше 900 мг (расчетная, оптимальная в каждом конкретном случае) наблюдалось достоверно резкое — в 1,5-2 раза снижение концентрации эстрадиола и прогестерона. При дозе менее 600 мг (расчетная, оптимальная) такого снижения не отмечено. Таким образом, из приведенных примеров проведения ТЛМ у больных разных возрастных групп, с разными патологиями можно сделать следующие выводы. 1. Используя данные ТЛМ, возраст и пол больного, а также эффективность проводимой терапии, проанализировав фармакокинетические параметры, полученные в результате измерения концентраций препарата в крови, на основе той или иной фармакокинетической модели, соответствующей вычисленным параметрам, можно составить оптимальный прогноз необходимого уровня препарата в крови больного при изменении режима дозирования. 2. Применяя современные подходы в оптимизации фармакотерапии, можно существенно повысить эффективность проводимой терапии и существенно сократить время пребывания в стационаре (а в некоторых случаях проводить лечение и амбулаторным путем). При этом значительно уменьшается число случаев резистентности, неясных побочных эффектов и общее число токсических проявлений. Методы определения антиконвульсантов (карбамазепин, фенитоин, фенобарбитал)Для определения концентрации антиконвульсантов в крови человека разработано достаточно большое количество методов, основанных на различных физико-химических свойствах этих препаратов. В настоящее время наиболее широко применяются иммуно-флюоресцентные и хроматографические методы. Различными авторами разработано большое количество газохроматографических методов и методов, основанных на применении высокоэффективной жидкостной хроматографии. На основе этих публикаций нами разработана оригинальная модификация метода определения концентрации антиконвульсантов с применением ВЭЖХ. Отличительная особенность разработанного нами метода заключается в одновременном определении карбамазепина, фенобарбитала, суксилепа и дифенина и з одного образца крови и в возможности применения микроколоночного варианта ВЭЖХ. Для получения максимальной чувствительности суксилепа необходимо проводить анализ при другой длине волны поглощения. В том случае, когда исследователь имеет дело с монотерапией вышеназванных антиконвульсантов, вполне возможно применять наиболее простой способ подготовки биологических проб для анализа — использовать метод прямого осаждения белков сыворотки крови. Осаждение белков может осуществляться, например, метиловым спиртом, 10%-ным раствором трихлоруксусной кислоты или ацетонитрилом с последующим центрифугированием и вводом надосадочной жидкости в хроматографическую колонку. В случае наличия в одной пробе двух и более препаратов наиболее целесообразно применение однократной экстракции. После отбора крови (1-2 мл) из локтевой вены ее выдерживают 30 мин. при комнатной температуре для образования сгустка, затем центрифугируют при 1500 об./мин. 15 мин при комнатной температуре. Полученную сыворотку крови замораживают (если не требуется проведения немедленного анализа) и хранят до проведения анализа при -20°С в морозильной камере. Анализ проводят методом ВЭЖХ с использованием однократной экстракции препарата хлористым метиленом или хлороформом и анализом сухого (после упаривания растворителя) на обратно-фазной хроматографической колонке на оптимальной длине волны спектрофотометрического детектора следующим образом. К 0,2 мл сыворотки крови, полученной описанным выше способом, приливали 1,5 мл хлористого метилена (возможно применение хлороформа, однако температура кипения хлористого метилена существенно ниже, что заметно влияет на скорость проведения всего анализа в целом). Смесь энергично встряхивали на вибромиксере 2 мин., центрифугировали при 5000 об./мин. при комнатной температуре 15 мин и затем отбирали 1,2 мл нижнего органического слоя. Для получения сухого остатка хлористый метилен упаривали при 50°С в токе азота. Сухой остаток растворяли в 120 мкл элюента и 50 мкл полученного раствора вводили в хроматографическую колонку. Хроматографическое разделение проводили при комнатной температуре (20+/-4°С) на колонке (4,6*250 мм) «Партисил ОДС-2» зернением 5 мкм. В качестве детектора использовали переменно-волновой спектрофотометр «Алтекс-Хитачи 155-40» с объемом ячейки 8 мкл на длине волны 254 нм. В качестве элюента использовалась смесь: 37% ацетонитрила и 63% бидистиллированной воды. Объемная скорость элюирования составляла 1,0 мл/мин. Время удерживания карбамазепина в этих условиях составляло 4,5+/-0,5 мин. Количественное определение препарата проводили по методу абсолютной калибровки. Для этого составляли контрольные растворы сыворотки крови, содержащие известные концентрации карбамазепина в диапазоне от 50 до 1000 нг/мл. Анализ каждого контрольного образца проводили не менее трех раз. По результатам проведенных определений составляли калибровочный график. Коэффициент корреляции r=0,998. Относительная ошибка определения карбамазепина при концентрации препарата в контрольном образце 2 мкг/мл составила 7,8% отн. Предел детектирования составлял 25 нг/мл. Аналогичным способом проводили анализы фенитоина и фенобарбитала. Вышеназванные препараты отличались только временем удерживания хроматографического пика и составляли 3,7+/-0,3 и 2,4+/-0,2 мин. соответственно. Существенное преимущество дает применение микроколоночного варианта ВЭЖХ. При использовании колонок с внутренним диаметром 2 мм и длиной 85-120 мм расход элюента уменьшается во много раз (например, в случае определения антиконвульсантов, расход элюента составлял 0,2 мл/мин), что существенно удешевляет стоимость проведения анализа, дает возможность использовать значительно меньший объем биологической пробы и увеличивает чувствительность определения препарата. Так, в нашей лаборатории разработаны методы определения антиконвульсантов с применением отечественных микроколонок, заполненных суспензионным способом отечественными носителями на основе «Диасорба С16» и других сорбентов. Типичная хроматограмма сывороточных экстрактов до приема препаратов и содержащих фепобарбитал, фе-нитоин и карбамазепин представлена на рис. 1. Рисунок 1. Хроматограммы сывороточных экстрактов до приема препаратов — I и содержащих фенобарбитал (Ф), фенитоин (D) и карбамазепин (К) — II Abstract One of the basic tasks of clinical pharmacokinetics is the maintenance of optimal concentration of a medicine in place of action. Especially it concerns preparations having narrow therapeutic corridor (some antibiotics, antidysrythmics, antiepyleptics etc.). This task is solved with the help Therapeutic Drug Monitoring (TDM). Fora lot of medicinal preparations the assignment of so-called average dozes without the account of knowledge of concentration of a preparation in blood can result in unpredictable consequences. What the basic principles of realization TDM are? After introduction of a preparation into organism its molecule at place of action should be in balance with molecules of same preparation in blood. It means, that optimal therapeutic effect should meet a certain average concentration (or average range of concentration) of preparation in blood. At a stage of development of new drugs it is mandatory to carry out pharmacokinetic studies allowing to find out optimum of «therapeutic corridor» for many medicines, which can be formulated as a range of concentration of a preparation in blood, within the limits of which there is a high enough probability of reception of a positive effect and low enough probability of occurrence undesirable and toxic effects.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Фармакокинетика, определение которой иногда сводят к воздействию организма на лекарственное вещество, описывает поступление препарата в организм, его трансформацию и выведение из организма; ключевыми этапами этого движения являются процессы абсорбции, биодоступности, распределения, метаболизма и экскреции.
Фармакодинамика, под которой понимают то, как препарат действует на организм, включает в себя связывание с рецептором, пострецепторные эффекты и химические взаимодействия.
Фармакокинетика ЛС определяет время начала, продолжительность и интенсивность вызываемого им эффекта. Формулы, математически описывающие эти процессы, помогают обобщить фармакокинетическое поведение большинства препаратов (см. таблицу Основные фармакокинетические параметры).
На фармакокинетику ЛС влияют как химические свойства препарата, так и индивидуальные особенности пациента. Некоторые из них (генотип, пол, возраст) могут быть использованы для прогнозирования фармакологического ответа у соответствующих категорий пациентов. Например, период полувыведения некоторых препаратов (особенно тех, выведение которых включает метаболизм и экскрецию) может существенно увеличиваться у пожилых пациентов (см. рисунок Сравнение фармакокинетического эффекта диазепама у молодого человека [A] и пожилого человека [B]). В действительности, возрастные физиологические изменения в организме влияют на многие аспекты фармакокинетики (см. Фармакокинетический эффект у людей страшего возраста и Фармакокинетический эффект у детей) (1, 2).
Другие факторы связаны с индивидуальной физиологией. Влияние некоторых индивидуальных факторов (например, почечной недостаточности, ожирения, печеночной недостаточности, обезвоживания) можно обоснованно предсказать, но другие факторы являются идиосинкразическими и, таким образом, приводят к непредсказуемым последствиям. Из-за индивидуальных особенностей введение лекарственного средства должно основываться на потребностях каждого пациента — традиционно, путем эмпирической корректировки дозы до достижения терапевтической цели. Такой подход часто является нецелесообразным, поскольку он может отсрочить оптимальный ответ или привести к нежелательным эффектам.
Знание принципов фармакокинетики может помочь врачу более точно и быстро провести коррекцию режима дозирования. Применение этого подхода в клинической практике получило название терапевтический лекарственный мониторинг.
Диазепам метаболизируется в печени додесметилдиазепама с участием ферментов цитохрома Р450. Десметилдиазепам – активное седативное средство, выводимое почками. 0-время введения. (Adapted from Greenblatt DJ, Allen MD, Harmatz JS, Shader RI: Diazepam disposition determinants. Clin Pharmacol Ther 27:301–312, 1980. doi: 10.1038/clpt.1980.40)
-
1. Le J, Bradley JS. Optimizing Antibiotic Drug Therapy in Pediatrics: Current State and Future Needs. J Clin Pharmacol 2018;58 Suppl 10:S108-S122. doi:10.1002/jcph.1128
-
2. Mangoni AA, Jackson SH. Age-related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol 2004;57(1):6-14. doi:10.1046/j.1365-2125.2003.02007.x
Лекарственная форма определяет способ доставки препарата, его стабильность и, конечно, фармакокинетические свойства. В этом отношении существенно выделяются средства с пролонгированным высвобождением. В чем их преимущества и почему такие препараты важно отпускать строго в том виде, который указан в рецепте? Рассмотрим на примере антидепрессанта третьего поколения венлафаксина1.
От общего – к частному
В терапевтической практике сегодня применяются как таблетки венлафаксина немедленного, неконтролируемого высвобождения, так и капсулы с пролонгированным действием.
Оба препарата имеют одни и те же фармакологические свойства. Венлафаксин взаимодействует с серотонинергическими и норадренергическими рецепторами, устраняя в нейрональных синапсах дефицит и серотонина, и норадреналина, что обеспечивает двойное действие и мощный эффект1,2, подтвержденный в метаанализе 34 рандомизированных двойных слепых исследований, включивших 8000 пациентов с депрессивными расстройствами. Его результат продемонстрировал статистически значимые преимущества венлафаксина по сравнению со всей группой СИОЗС*, в которую входит циталопрам, эсциталопрам, флуоксетин, флувоксамин, пароксетин и сертралин1.
Будучи идентичными по фармакологической активности, капсулы и таблетки антидепрессанта третьего поколения существенно отличаются фармакокинетически, что определяет их переносимость и кратность применения.
Ключевой момент
Таблетки, высвобождение активного вещества из которых не контролируется, имеют относительно небольшой период полувыведения – 5 часов для венлафаксина и 11 часов для основного активного метаболита2 (у оригинального препарата Велаксин® в таблетированной форме).
А вот капсулы с пролонгированным действием обеспечивают более стабильную концентрацию активного компонента в крови в течение суток и имеют менее выраженный пик концентрации в крови после приема1,3.
Капсулированная форма Велаксин® по сравнению с таблетированной1–4:
- проявляет менее выраженные побочные эффекты, особенно на пике концентрации
- имеет менее выраженный и позже развивающийся синдром отмены
- после случайного пропуска или опоздания в приеме венлафаксина с пролонгированным действием вероятность развития синдрома отмены намного меньше по сравнению с препаратом в обычной форме4
- имеет однократный режим применения, в то время как таблетки принимаются строго 2 раза в день.

Свойства капсул Велаксин® обеспечивают большую приверженность к лечению по сравнению с таблетками1,4, а значит, и оптимальный терапевтический ответ. И у пациента есть возможность его получить, если первостольник, обслуживая рецепт, отпустит препарат в форме, в которой его выписал врач, – Велаксин® капсулы.
3 шага рецептурного отпуска Велаксин®


Форма определяет свойства
| Таблетки Велаксин® | Капсулы Велаксин® | |
|---|---|---|
| Особенности лекарственной формы | Твердая форма, полученная прессованием порошка |
Смесь пеллет, заключенных в желатиновую капсулу |
| Высвобождение действующего вещества | Неконтролируемое | Модифицированное |
| Максимальная концентрация в плазме | В течение 2,4 ч, основной метаболит – через 4,3 ч после введения2 | Венлафаксин – в течение 6,0 ± 1,5 ч, основной активный метаболит – 8,8 ± 2,2 ч3 |
| Период полувыведения | Венлафаксин – 5 ч, основной активный метаболит – 11 ч2 | Венлафаксин – 15 ± 6 ч3 |
Благодаря лекарственной форме Велаксин® капсулы обеспечивают1,4:
• более стабильную концентрацию венлафаксина в крови в течение суток
• менее выраженный пик концентрации
Пролонгированные формы венлафаксина больше подходят для лечения депрессии, в том числе с тревожным компонентом, чем обычные1,4
Венлафаксин пролонгированного действия, в отличие от таблеток, не вызывает обострения тревоги на пике концентрации в крови.
Поэтому FDA** одобрило применение1,4:
капсулы пролонгированного действия – для лечения депрессивных расстройств с тревожным компонентом
таблетки – только для лечения депрессивных расстройств

Фармакокинетические особенности препарата Велаксин® капсулы обусловливают их преимущества по сравнению с таблетками по ряду параметров1,4
Переносимость1,4
• менее выраженные побочные эффекты
• менее выраженный и позже развивающийся синдром отмены
• более низкий риск развития синдрома отмены после случайного пропуска или опоздания в приеме
Удобство применения2,3
• Капсулы применяются однократно в сутки (таблетки – строго 2 раза в день)
Приверженность терапии1,4
• При однократном режиме применения, по сравнению с приемом 2 раза в день, приверженность к лечению выше в разрезе как кратности приема, так и длительности терапии5***
Эффективность
• По данным исследований, пролонгированная форма венлафаксина статистически достоверно эффективнее по сравнению с препаратом немедленного высвобождения6,7
Капсулы/таблетки в вопросах и ответах
Почему некорректно заменять капсулы венлафаксина на таблетки?
Капсулы и таблетки венлафаксина существенно разнятся по фармакокинетическим параметрам, что определяет их отличия в переносимости, режиме дозирования и даже эффективности1,4,6-7. Именно поэтому отпускать препарат следует в форме, указанной в рецепте.
Как объяснить клиенту, почему нужно принимать именно капсулы венлафаксина, если их выписал врач?
Врач, назначая капсулы, исходил из их преимуществ по сравнению с таблетками. Так, капсулы Велаксин® лучше переносятся, имеют менее выраженный синдром отмены, в том числе и после случайного пропуска или опоздания в приеме, и принимаются один раз в день, а не два, как таблетированная форма1,4.
* Селективные ингибиторы обратного захвата серотонина.
** Управление по контролю пищевых продуктов и лекарств в США.
*** При хронических заболеваниях4.
1. Пантелеева Г.П. и соавт. Лечение эндогенных депрессий венлафаксином: клиническое действие, переносимость и персонифицированные показания к назначению // Журнал неврологии и психиатрии им. C.C. Корсакова, 2015. Т. 115. № 1–2. С. 43–51.
2. Инструкция по медицинскому применению лекарственного препарата Велаксин® таблетки. Рег. уд. №: ЛС–000318 от 15.03.2010.
3. Инструкция по медицинскому применению лекарственного препарата Велаксин® капсулы. Рег. уд. №: ЛСР–000030 от 31.05.2007.
4. Беккер Р.А., Быков Ю.В. Велаксин® в капсулах: представление новой пролонгированной формы и обзор новейших данных об эффективности и безопасности // Психиатрия и психофармакотерапия, 2017. Т. 19. № 6. С. 33–41.
5. Coleman C. I. et al. Dosing frequency and medication adherence in chronic disease // Journal of Managed Care Pharmacy. 2012; 18 (7): 527–539.
6. Entsuah R., Chitra R. A benefit-risk analysis of once-daily venlafaxine extended release (XR) and venlafaxine immediate release (IR) in outpatients with major depression // Psychopharmacology bulletin. 1997; 33 (4): 671–676.
7. Cunningham L. A. Once-daily venlafaxine extended release (XR) and venlafaxine immediate release (IR) in outpatients with major depression // Annals of clinical psychiatry. 1997; 9 (3): 157–164.
Фармакокинетика — раздел клинической фармакологии, изучающий пути введения, биотрансформацию, связь с белками крови, распределение и выведение лекарственных средств (ЛС).
Один из основных показателей, определяющих фармакологический эффект, — концентрация ЛС в области рецептора, однако в условиях целостного организма установить её невозможно. Экспериментально доказано, что в большинстве случаев имеется корреляция между концентрацией препарата в крови и его содержанием в других биологических жидкостях и тканях.
Поэтому для определения фармакокинетических параметров ЛС изучают его содержание в крови. Чтобы получить соответствующие представления о поступлении препарата в кровь и выведении его из организма, определяют содержание ЛС в плазме крови в течение длительного времени, используя методы жидкостной или газожидкостной хроматографии, радиоиммунный и иммуноферментный анализы, спектрофотометрический метод. На основании полученных данных строят график (фармакокинетическую кривую), отмечая на оси абсцисс время исследования, а на оси ординат — концентрацию ЛС в плазме крови.
В связи со сложностью описания деталей процесса распределения ЛС во всех органах и тканях, организм условно представляют в виде одной или нескольких изолированных проницаемой мембраной частей (камер), в которых Л С распределяется. Этот вид моделирования называют камерным. За центральную камеру обычно принимают кровь и хорошо кровоснабжаемые органы (сердце, лёгкие, печень, почки, эндокринные железы), за периферическую — менее интенсивно кровоснабжаемые органы и ткани (мышцы, кожу, жировую ткань). В этих камерах ЛС распределяется с разной скоростью: быстрее — в центральной, медленнее — в периферической. К наиболее простым относят однокамерную модель, когда предполагают, что после введения препарата его концентрация убывает по моноэкспоненциальному закону. В соответствии с законами линейной кинетики скорость изменения количества препарата в камере пропорциональна его количеству в этой камере.
Кажущийся объём распределения (Vd) — гипотетический объём жидкости организма, необходимый для равномерного распределения всего количества ЛС (введённой дозы) в концентрации, аналогичной таковой в плазме крови. Этот показатель измеряют в л/кг. При внутривенном введении объём распределения равен отношению дозы ЛС к его начальной концентрации в крови.
• Высокие значения объёма распределения свидетельствуют о том, что ЛС активно проникает в биологические жидкости и ткани. При этом, если ЛС активно связывается, например, жировой тканью, его концентрация в крови может практически мгновенно стать очень низкой, а объём распределения достигнет нескольких сотен литров, превысив реальный объём жидкостей организма. Поэтому этот показатель и называют кажущимся объёмом распределения.
• Объём распределения зависит от различных факторов.
— Физико-химические свойства ЛС (молекулярная масса, степень ионизации и полярности, растворимость в воде и жирах) влияют на его прохождение через мембраны.
— Физиологические факторы (возраст, пол, общее количество жировой ткани в организме). Например, у пожилых людей и ново рождённых Vd снижен.
— Патологические состояния, особенно заболевания печени, почек, сердечно-сосудистой системы (ССС).
Максимальная концентрация (Сmax) и время наступления максимальной концентрации (Тmax). При поступлении ЛС в системный кровоток (в случае внесосудистого введения) его концентрация постепенно возрастает, достигая значения (Сmax) в момент Тmax, а затем начинает снижаться.
• Если процесс абсорбции имеет линейный характер (скорость процесса прямо пропорциональна количеству ЛС в системе), скорость этого процесса характеризуется константой абсорбции (kabs), измеряемой в часах и рассчитывается через период полувсасывания (Т1/2abs) — время, в течение которого всасывается 1/2 введённой дозы препарата.
Биодоступность (F) — часть дозы Л С (в %), достигшая системного кровотока после вне-сосудистого введения (в этом случае не всё количество препарата достигает системного кровотока).
• Абсолютную биодоступность определяют соотношением значений площади под кинетической кривой (area under curve, AUC) при вне-сосудистом и внутривенном введениях препарата.
— В рамках однокамерной модели при внутривенном введении площадь под кинетической кривой определяется отношением начальной концентрации в крови (Со) к константе элиминации (кеl)
AUC = C0/kel
— AUC прямо пропорциональна однократной дозе ЛС, введённой внутривенно (в/в), и обратно пропорциональна общему клиренсу препарата. Она связана с величиной объёма распределения:
Vd=D/kel·AUC,
где Vd — объём распределения, кеl — константа элиминации, D — доза, AUC — площадь под кинетической кривой.
• Биоэквивалентность (относительная биодоступность) — соотношение количества ЛС, поступившего в системное кровообращение при применении его в различных лекарственных формах или лекарственных препаратах, выпускаемых различными фирмами. Если сравниваемые ЛС аналогичны (действующее вещество, доза, лекарственная t форма), но изготовлены разными производителями, их называют дженериками, и в этом случае необходимо исследование их биоэкви— валентности. Два лекарственных препарата биоэквивалентны, если они обеспечивают одинаковую биодоступность ЛС.
Константа скорости элиминации (кеl) — процент снижения концентрации вещества в крови в единицу времени (отражает долю препарата, выводимую из организма за единицу времени). Элиминация складывается из процессов биотрансформации и экскреции. Константа скорости элиминации характеризует элиминацию в рамках однокамерной модели при линейном характере процесса выведения. Период полувыведения (Т1/2) — время, необходимое для снижения концентрации препарата в крови на 50% в результате элиминации. В рамках линейной модели Т1/2 рассчитывают по формуле:
Т1/2 =0,693/Kel
• Практически за один Т1/2 из организма выводится 50% ЛС, за два периода — 75%, за 3 периода — приблизительно 90% и т.д.
• Зависимость между Т1/2 и кеl важна для подбора режима дозирования и особенно для определения интервала между дозами.
Клиренс (CI) — объём плазмы или крови, полностью освобождающийся от ЛС в единицу времени. Этот показатель количественно характеризует выведение препарата и выражается в мл/мин или л/ч. В рамках линейной модели клиренс рассчитывают по формуле:
Cl=Vd·Kel=D/AUC
где Сl — клиренс, Vd — объём распределения, Ке1 — константа скорости элиминации, D — доза, AUC — площадь под кинетической кривой.
• Общий клиренс представляет собой сумму почечного и печёночного клиренсов (так как эти органы служат основными путями выведения ЛС). (Другие пути выведения или внепечёночный метаболизм при расчёте общего клиренса обычно не учитывают.)
— Печёночный клиренс характеризует биотрансформацию ЛС в печени (метаболический клиренс) и выведение с жёлчью (жёлчный клиренс).
— Почечный клиренс отражает выведение препарата с мочой. На пример, почечный клиренс циметидина приблизительно составляет 600 мл/мин, метаболический — 200 мл/мин, жёлчный — 10 мл/мин, поэтому общий клиренс равен 810 мл/мин.
• Основные физиологические факторы, определяющие клиренс, — функциональное состояние основных физиологических систем организма, объём притекающей крови и скорость кровотока в органе. Печёночный клиренс зависит от скорости печёночного кровотока или функциональной способности метаболизирующих ферментов. Например, клиренс лидокаина, интенсивно метаболизируемого печёночными ферментами, зависит прежде всего от скорости его доставки к печени (т.е. от объёма притекающей крови и скорости кровотока), поэтому, например, при застойной сердечной недостаточности он снижен. Клиренс же фенотиазинов зависит в основном от активности метаболизирующих ферментов, поэтому при поражении гепатоцитов клиренс препаратов этой группы резко снижается, вследствие чего концентрация их в крови значительно возрастает.
Равновесная (или стационарная) концентрация (Css) — концентрация, достигнутая при состоянии, когда в каждом интервале между приёмом очередных доз количество всасывающегося ЛС равно количеству элиминируемого [т.е. при стационарном (steady state), или равновесном, состоянии]. Т.е. если ЛС вводят в постоянной дозе через фиксированные интервалы времени, продолжительность которых меньше времени элиминации, его концентрация в крови возрастает, а затем колеблется в пределах средней величины между максимальными и минимальными значениями.
• При достижении С проявляется в полном объёме клинический эффект ЛС. Чем меньше Т1/2 ЛС, тем скорее достигается Си и тем выражение будут её колебания. Например, Т1/2 новокаинамида равен 2— 3 ч, и при назначении через каждые 6 ч его Css характеризуется большим разбросом значений. Поэтому для предупреждения и уменьшения колебаний Css в крови всё большее распространение получают лекарственные формы с замедленным высвобождением активного вещества.
В клинической практике фармакокинетические параметры используют, в частности, для расчёта назначаемых доз препаратов.
• Для расчёта нагрузочной дозы, требуемой для достижения необходимой эффективной концентрации ЛС в крови, используют объём распределения:
Dнагр=Vd·C
где Dнагр — нагрузочная доза, VD — объём распределения, С — концентрация ЛС в плазме крови.
• Для расчёта поддерживающей дозы, т.е. дозы, необходимой для поддержания нужной концентрации ЛС в крови, используют значение клиренса:
Dпод=Cl·Css
где Dnoд — поддерживающая доза, Сl — общий клиренс, Сss — равновесная концентрация.
К основным фармакокинетическим процессам относят всасывание, метаболизм (биотрансформацию), распределение и выведение ЛС.